Introduction
Elevation of blood cholesterol has been conclusively established in epidemiological studies as a major independent risk factor for coronary artery disease (CAD) and other atherosclerotic vascular diseases (ASCVD)1. Atherosclerosis is a lifelong disease process that commences in childhood and results in clinical disease in middle age onwards. It first appears as deposits of lipid in the intima of large muscular and elastic arteries, and progresses through several stages involving increased lipid deposition, smooth-muscle proliferation, scarring, vascularisation, haemorrhage, and eventually thrombosis before finally causing clinically evident ischaemic disease2. The relationship between low-density lipoprotein cholesterol (LDL-C) and CAD has been well established3. The causative role of LDL has been derived from three sources: genetic, in which mutations that led to impairment of the receptor-mediated removal of LDL from plasma cause fulminant atherosclerosis; experimental, in which animals with low levels of LDL have no atherosclerosis; and epidemiologic, in which humans with low LDL levels have very little atherosclerosis and the increment in the disease has been found to be proportional to LDL levels4. In the early stages of atherosclerosis, coronary arteries compensate for the accumulation of atherosclerotic plaque by enlarging or dilating the external elastic lamina. Once this compensatory dilatation reaches its limit, which is generally with plaque >40% of the reference cross-sectional area, lumen narrowing ensues5. Stable advance lesions usually have uniformly dense fibrous caps, whereas ones that are potentially dangerous are often non-occlusive but contain an abundance of macrophages. When macrophages enter, accumulate, and are activated at the shoulders of unstable lesions, erosion and rupture of the fibrous cap then leads to myocardial infarction (MI)6. Percutaneous coronary intervention (PCI) has been established as a definitive revascularisation method for patients with CAD, whether it be in the setting of a stable CAD or an acute coronary syndrome (ACS). Optimal lipid control post-PCI is of paramount importance to reduce further coronary-related events, given that the process of atherosclerosis is, in fact, a continuum7. Contemporary dyslipidaemia guidelines89 have categorised post-PCI patients under the very high-risk category of developing future cardiovascular (CV) events and it is imperative that post-PCI patients receive optimal secondary prevention. This review aims to provide an overview of guideline recommendations for secondary prevention of dyslipidaemia in post-PCI patients, the evidence and rationale behind treatment measures, the real-world issues and challenges faced in the implementation of secondary prevention, and the potential methods to overcome them.
Guidelines
The 2018 American College of Cardiology/American Heart Association (ACC/AHA) Guideline on the Management of Blood Cholesterol states that high-intensity statins that have the potential to lower LDL-C by ≥50% (either 40 to 80 mg daily of atorvastatin or 20 to 40 mg daily of rosuvastatin) should be initiated or continued with the objective of achieving a 50% reduction in LDL-C in patients with clinical ASCVD who are 75 years of age or younger. In patients older than 75, the evaluation of the potential for reduction of ASCVD risk, drug-drug interactions, adverse effects, patient frailty, and patient preference should be carried out. After this evaluation, either moderate-intensity statins (such as 10 to 20 mg daily of atorvastatin, 5 to 10 mg daily of rosuvastatin or 20 to 40 mg daily of simvastatin) or high-intensity statins can be initiated. Ezetimibe may be added should these patients continue to have an LDL-C level of ≥70 mg/dL (1.8 mmol/L) and they are already on maximally tolerated statin therapy. Should combination therapy of a high-intensity statin plus ezetimibe be insufficient to achieve LDL-C target, then the addition of a proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor can be considered8.
The 2019 European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) Guidelines for the management of dyslipidaemias are similar to the 2018 ACC/AHA Guidelines in terms of the treatment algorithm, except that the 2019 ESC/EAS Guidelines recommend a lower target of LDL-C of <55 mg/dL (1.4 mmol/L) that needs to be achieved in patients requiring secondary prevention for established ASCVD. LDL-C is recommended as the primary lipid parameter for screening, diagnosis, and management. Should a patient have recurrent cardiovascular (CV) events within 2 years while being on maximally tolerated statin therapy, an LDL-C target of <40 mg/dL (1.0 mmol/L) is recommended9.
Both sets of guidelines reinforce the importance of therapeutic lifestyle change (avoidance of cigarette smoking, having a healthy diet, achieving a good body weight, and undergoing adequate physical activity) in addition to optimisation of other concomitant CV risk factors89.
Rationale for statin therapy
In the Cholesterol Treatment Trialists’ (CTT) meta-analysis of individual participant data (IPD) from 169,138 participants in 26 randomised controlled trials (RCTs) of a statin versus control, or a more versus less intensive statin regimen, it was shown that for each 1 mmol/L reduction in LDL-C, there was a reduction of major vascular events (MI, CAD death, or any stroke or coronary revascularisation) by 22%, major coronary events by 23%, CAD death by 20%, and all-cause mortality by 10% over a period of 5 years10. Statins are lipid-lowering drugs that are highly effective in reducing circulating LDL-C and the risk of acute CV events. Through competitive inhibition on the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), statins reduce intrahepatic synthesis of cholesterol in addition to promoting nuclear translocation of transcription factor sterol regulatory element-binding protein 2 (SREBP-2)11. Implantation of a coronary stent leads to mechanical disruption of the vessel wall and mechanical injury which can cause distal embolisation of plaque microdebris, endothelial denudation, medial dissection, and exposure of subintimal components to inflammatory mediators, resulting in the activation of platelets, complements and coagulation12. Statins have been shown to stabilise plaque, improve endothelial function, inhibit platelet aggregation, and reduce inflammation13. In the post-stenting period, statin therapy is expected to reduce vascular, myocardial and systemic inflammation, reduce periprocedural infarction, and improve endothelial function within a period of hours to days. In days to weeks, statin therapy is expected to reduce inflammation and thrombosis, and inhibit vascular smooth muscle proliferation. In months to years, it is expected to prevent progression of CAD, stabilise atherosclerotic plaque and prevent MIs12.
The PROVE IT-TIMI 22 trial enrolled 4,162 patients who had ACS within the preceding 10 days and randomised them to either intensive statin therapy with 80 mg daily of atorvastatin or standard statin therapy with 40 mg daily of pravastatin. Intensive therapy with high-dose atorvastatin resulted in a median LDL-C level of 62 mg/dL (1.60 mmol/L) as compared with a level of 95 mg/dL (2.46 mmol/L) for standard-dose pravastatin14. Of the overall cohort, 2,868 (68.9%) patients underwent PCI for the index ACS prior to randomisation. Patients who received 80 mg daily of atorvastatin had a 22% relative risk (RR) reduction (hazard ratio [HR]: 0.78, 95% confidence interval [CI]: 0.67-0.91; p=0.001) of the 2-year Kaplan-Meier estimated event rate for the primary composite endpoint (death from any cause, MI, documented unstable angina [UA] requiring rehospitalisation and revascularisation [PCI or coronary artery bypass grafting {CABG} at least 30 days after randomisation] and stroke) compared to patients who received 40 mg daily of pravastatin (21.5% vs 26.5%). In addition, patients who were on atorvastatin also had a lower rate of target vessel revascularisation (TVR; 11.4% vs 15.4%, odds ratio [OR]: 0.73, 95% CI: 0.59-0.89, p<0.001) and non-TVR (8.0% vs 10.5%, OR: 0.75, 95% CI: 0.59-0.95, p=0.017). There were no significant differences in terms of safety between the groups apart from a higher incidence of liver-related side effects with high-dose atorvastatin15. In a substudy of the trial evaluating the safety and efficacy of achieving very low LDL levels with intensive statin therapy, there were no trends toward increases in the rates of expected side effects such as myopathy or elevations in liver enzyme levels based on achieved LDL levels. There was also no increase in intracranial haemorrhage, ophthalmologic side effects, trauma/suicide, or all-cause mortality when lower LDL levels were achieved. Patients who achieved very low LDL levels with intensive statin therapy were also found to have had fewer CV events16.
The Treating to New Targets (TNT) trial enrolled 10,001 patients with clinically evident CAD defined as at least one of the following: previous MI, previous or current angina with objective evidence of atherosclerotic CAD, and a history of coronary revascularisation. Patients were randomised to either 10 mg daily or 80 mg daily of atorvastatin and were followed up for a median of 4.9 years. Mean LDL-C levels during the study were 77 mg/dL (2.0 mmol/L) among patients receiving 80 mg of atorvastatin and 101 mg/dL (2.6 mmol/L) among those receiving 10 mg of atorvastatin17. In a post hoc analysis of the trial involving 5,407 patients who had a previous PCI, patients who were randomised to 80 mg daily of atorvastatin had a 21% RR reduction and a 2.1% absolute risk reduction (HR: 0.79, 95% CI: 0.67-0.94, p=0.008) of the primary outcome (time to the first occurrence of a major CV event defined as: death from CAD, non-fatal non-procedure-related MI, resuscitated cardiac arrest, and fatal or non-fatal stroke) compared to patients randomised to 10 mg daily of atorvastatin. Patients in the high-dose atorvastatin group were also less likely to undergo repeat coronary revascularisation, either PCI or CABG, compared to patients in the low-dose group (27% RR reduction and a 5.6% absolute risk reduction [HR: 0.73, 95% CI: 0.65-0.82, p<0.0001]). Of note, there was a significantly higher incidence of transaminitis amongst patients receiving 80 mg of atorvastatin18.
In a meta-analysis of CV outcome trials (with a total of 27,548 patients) comparing intensive versus moderate statin therapy, there was a significant odds reduction of 16% for coronary death or MI (9.4% vs 8.0%, OR: 0.84, 95% CI: 0.77-0.91, p<0.00001) along with a significant 16% reduction of coronary death or any CV events (32.3% vs 28.8%, OR: 0.84, 95% CI: 0.80-0.89, p<0.0000001). An extrapolation of these findings would mean that for every million patients with CAD, intensive statin therapy would prevent more than 35,000 CV events (including more than 14,000 coronary deaths or MIs) should they be treated for 5 years, as compared to standard-dose statin therapy. This produces a number needed to treat of only 29 patients (for 2 years following an ACS, or for 5 years in patients with stable CAD) to prevent a CV event19.
In the prospective REAL-CAD trial, 13,054 Japanese patients with stable CAD who achieved LDL-C of <120 mg/dL were randomised to either 4 mg or 1 mg daily of pitavastatin. A total of 79.4% of patients had undergone prior PCI. After a median follow-up of 3.9 years, high-dose pitavastatin significantly reduced the risk of the primary endpoint (composite of CV death, non-fatal MI, non-fatal ischaemic stroke, or UA requiring emergency hospitalisation) (4.3% vs 5.4%, HR: 0.81, 95% CI: 0.69-0.95, p=0.01) and the risk of the secondary composite endpoint (a composite of the primary endpoint and clinically indicated coronary revascularisation excluding target lesion revascularisation [TLR] at sites of prior PCI; 7.9% vs 9.7%, HR: 0.83, 95% CI: 0.73-0.93, p=0.002) as compared to low-dose pitavastatin. During the entire course of follow-up, LDL-C in the high-dose group was lower by 14.7 mg/dL than in the low-dose group (p<0.001). The rates of serious adverse events were low and did not differ between the 2 groups20.
In the ESTABLISH study, patients who underwent PCI for ACS were randomised to receive either 20 mg daily of atorvastatin or placebo post-PCI. After 6 months, LDL-C level was significantly decreased by 41.7% in the atorvastatin group compared with an increase of 0.7% in the control group (p<0.0001). Plaque volume as assessed by intravascular ultrasound (IVUS) was significantly reduced in the atorvastatin group (13.1±12.8% decrease) compared with the control group (8.7±14.9% increase, p<0.0001). There was a significant positive correlation between percent change in plaque volume with follow-up LDL-C level (r=0.456, p=0.0011) and percent LDL-C reduction (r=0.612, p<0.0001)21.
The YELLOW trial randomised 87 patients with stable and multivessel CAD to 40 mg daily of rosuvastatin (intensive) or standard lipid-lowering therapy after successful PCI of the target lesion. Non-target lesions with >70% diameter stenosis and a fractional flow reserve (FFR) of ≤0.80 were further evaluated with IVUS and near-infrared spectroscopy (NIRS). After 6 to 8 weeks, repeat coronary angiography, FFR, IVUS, and NIRS were performed at the same non-target lesion previously imaged. Median follow-up was 50 days. At the end of follow-up, LDL-C levels of 58.4±26.3 mg/dL in the intensive group were significantly lower compared to 81.9±27.9 mg/dL in the standard group after treatment (p<0.001). Patients randomised to rosuvastatin had a higher lipid-core burden index (LCBI)4mm max (p=0.01) and lesion LCBI (p=0.04) at baseline compared with the standard group. At the end of follow-up, significant reductions in LCBI were only seen in the intensive group. LCBI4mm max decreased from 490.6 (363.8-689.7) at baseline to 336.1 (252.3-479.9) at follow-up (p=0.01). LCBI at the lesion was also reduced in the intensive group, from 132.4 (99.0-201.2) at baseline to 99.8 (64.2-159.3) at follow-up (p=0.02)22.
In a post hoc patient-level analysis of 8 prospective randomised trials using serial IVUS, patients with CAD receiving high-intensity statin therapy were found to have a regression of percent atheroma volume (PAV) from baseline (-0.6±0.1%, p<0.001), while patients receiving low-intensity statin therapy and no-statin therapy were found to have a progression of PAV (+0.8±0.1% and +1.0±0.1%, p<0.001, respectively). Statins were also found to promote calcification of coronary atheroma (patients who were on high-intensity statin therapy had the greatest increase in calcium), and statin-mediated atheroma calcification may improve plaque stability following long-term usage of high-intensity statin therapy23.
Rationale for ezetimibe therapy
Ezetimibe targets the Niemann–Pick C1–like 1 (NPC1L1) protein; it acts at the brush border of the small intestine and inhibits the uptake of dietary and biliary cholesterol into the enterocytes24. In a study involving 628 patients with baseline LDL-C of 145 to 250 mg/dL, 10 mg daily of ezetimibe, when added to atorvastatin, provided a significant additional 12% LDL-C reduction which yielded total LDL-C reductions of 50% to 60%. LDL-C reductions with ezetimibe plus 10 mg daily of atorvastatin were similar compared to 80 mg daily of atorvastatin alone. The combination was well tolerated, with a safety profile similar to atorvastatin alone and to placebo25.
In the IMPROVE-IT trial, 18,144 patients who were hospitalised for an ACS, and had LDL-C levels of 50 to 100 mg/dL (1.3 to 2.6 mmol/L) if they were receiving lipid-lowering therapy or 50 to 125 mg/dL (1.3 to 3.2 mmol/L) if they were not receiving lipid-lowering therapy, were randomised to receive either 40 mg daily of simvastatin plus 10 mg daily of ezetimibe or 40 mg daily of simvastatin alone. A total of 70% of patients underwent PCI prior to randomisation. At 7 years, patients in the combination therapy group had a lower rate (32.7%) of the primary endpoint (composite of CV death, non-fatal MI, UA requiring rehospitalisation, coronary revascularisation [≥30 days after randomisation], or non-fatal stroke) as compared to the monotherapy group (34.7%; HR: 0.936, 95% CI: 0.89-0.99, p=0.016). Rates of prespecified treatment-related adverse events were similar between both groups26.
Rationale for PCSK9 inhibitor therapy
PCSK9 is a serine protease encoded by a gene comprising 12 exons, located on chromosome 1p32.3. It is secreted from liver cells, circulates in the plasma, binds to an LDL receptor, and is subsequently internalised together with the LDL receptor, thereby promoting the cellular degradation of the receptor27. PCSK9 inhibitors inhibit its effects on LDL receptors, resulting in a reduction of LDL-C28. On its own, a PCSK9 inhibitor reduces LDL-C by ≈60%. When combined with a high-intensity statin, an LDL-C reduction of ≈75% is achieved and when both are combined with ezetimibe an LDL-C reduction of ≈85% is achieved9.
In the FOURIER trial, 27,564 patients who were receiving statin therapy and had established ASCVD and LDL-C levels of 70 mg/dL (1.8 mmol/L) or higher were randomised to receive evolocumab (either 140 mg every 2 weeks or 420 mg monthly) versus placebo. The median duration of follow-up was 2.2 years. At 48 weeks, patients who received evolocumab had a significantly lower risk of the primary endpoint (composite of CV death, MI, stroke, hospitalisation for UA, or coronary revascularisation; 9.8% vs 11.3%, HR: 0.85, 95% CI: 0.79-0.92, p<0.001) compared to patients in the placebo group. Apart from a nominally higher rate of injection-site reaction in the evolocumab group, the rates of other adverse events were similar between both groups29. In a separate analysis of FOURIER, as compared to placebo, evolocumab significantly reduced the risk of simple PCI by 22% (HR: 0.78, 95% CI: 0.70-0.88, p<0.001) and complex PCI by 33% (HR: 0.67, 95% CI: 0.54-0.84, p<0.001) in 1,482 patients who underwent PCI during the follow-up period30.
In a retrospective, non-randomised, observational, single-centre study, 64 patients with multivessel disease and untreated dyslipidaemia who underwent PCI for ACS were randomised to receive 5 mg daily of rosuvastatin or evolocumab 140 mg every 2 weeks on top of 5 mg daily of rosuvastatin. At 12-week follow-up there was a significant increment in the minimum fibrous cap thickness (177.7±33.2 vs 164.0±30.4 μm, p<0.001) and a significant reduction of the lipid arc (96.2±37.0 vs 110.8±39.7 degrees, p<0.001) on optical coherence tomography (OCT) in the combined evolocumab and rosuvastatin group31.
In the ODYSSEY OUTCOMES trial, 18,924 patients who had an ACS 1 to 12 months prior, had an LDL-C level of at least 70 mg/dL (1.8 mmol/L) and were receiving statin therapy at a high-intensity dose or at the maximum tolerated dose, were randomised to receive either 75 mg every 2 weeks of alirocumab or placebo. The median duration of follow-up was 2.8 years and 72.3% of patients underwent PCI or CABG for the index ACS episode. The alirocumab group had a lower rate (9.5%) of the primary endpoint (composite of death from coronary heart disease, non-fatal MI, fatal or non-fatal ischaemic stroke, or UA requiring hospitalisation) compared to the placebo group (11.1%; HR: 0.85, 95% CI: 0.78-0.93, p<0.001). The rates of adverse events were similar between both groups apart from a slightly higher rate of injection-site reaction in the alirocumab group32.
Real-world challenges and issues
In the real-world setting, lipid goal attainment has been suboptimal globally. In a retrospective analysis of 42,767 German patients at high or very-high CV risk, only 35% of patients received statins, and most statin-treated patients (32.2%) were receiving low-to-moderate intensity statins. Additionally, LDL-C threshold attainment was low among patients receiving lipid-lowering therapy, with only 13.5% achieving a level of <70 mg/dL (1.8 mmol/L) and 36.8% achieving a level of 70 to <100 mg/dL (1.8 to <2.6 mmol/L)33. In the ESC-EORP EUROASPIRE V survey, it was found that the overall lipid control among 7,824 patients with underlying CAD was not satisfactory where there were elevated levels of total cholesterol (TC), LDL-C, non-high-density lipoprotein cholesterol (HDL-C) and triglyceride (TG), and low levels of HDL-C. Only 55% of patients were discharged on a high-intensity statin and only 2.7% were on a combination of a high-intensity statin with ezetimibe at the time of interview34.
In the DYSIS-II Europe study, 4,344 patients were enrolled, of whom 2,946 had stable CAD and 1,398 were admitted for an ACS. Among the stable CAD patients, 57.1% had prior PCI and only 28.3% achieved the LDL-C target of <70 mg/dL. Only 15.7% of patients with ACS achieved the necessary LDL-C target. Use of non-statin lipid-lowering therapy was noted to be low, with only 11.6% of the stable CAD patients being treated with a combination of a statin and ezetimibe, and 6.4% and 5.7% of the ACS patients being treated with this combination at hospital admission and follow-up, respectively35.
In addition to statin underutilisation, non-adherence to lipid-lowering therapy for secondary prevention remains an important obstacle to event reduction of CAD. Although statins are generally well-tolerated, statin adherence is poor in clinical practice36. Patients may be fearful of the potential adverse effects of statins, but this fear is often triggered and perpetuated by reports published and circulated via social media which often lack a scientific basis37. The only adverse events that have been reliably shown to be caused by statin therapy are myopathy, new-onset diabetes mellitus, and a probable increase in haemorrhagic strokes. Treatment of 10,000 patients for 5 years with a standard statin regimen would be expected to cause about 5 cases of myopathy, 50 to 100 new cases of diabetes mellitus, and 5 to 10 haemorrhagic strokes38.
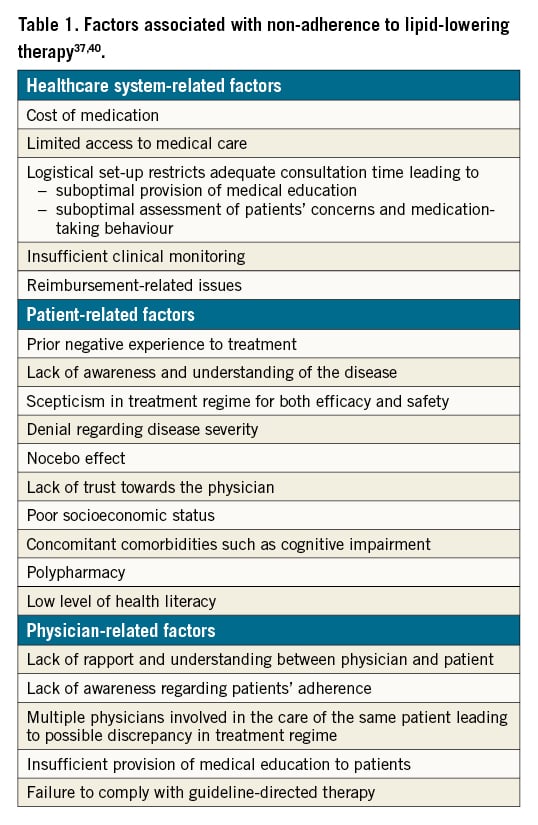
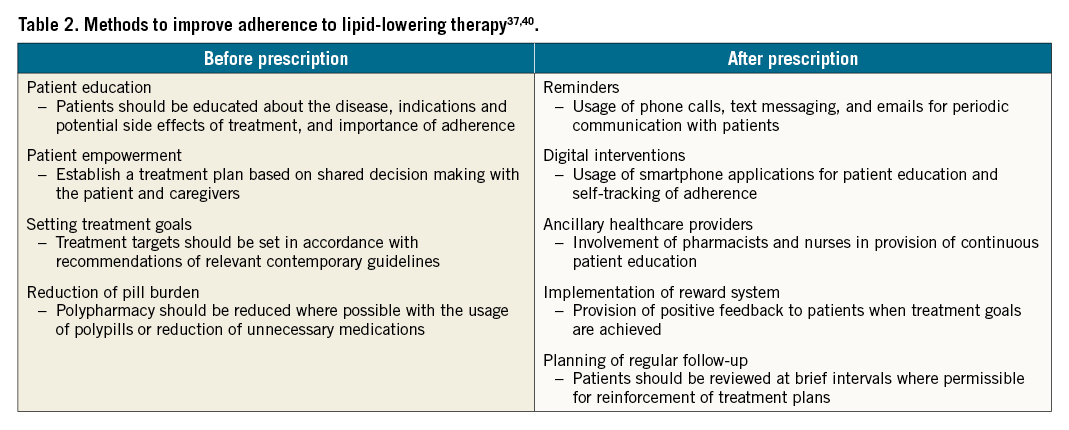
Good adherence to guideline-recommended statin therapy has been proven to be associated with an improved outcome39. Adherence refers to the extent to which a patient’s medication-taking practice coincides with prescribed medical recommendations. Although many patient characteristics can affect statin efficacy, non-adherence is among the most important determinants of outcome40. As stated in Table 1, factors associated with non-adherence to lipid-lowering therapy can be divided into healthcare system-related, patient-related and physician-related. The various methods for optimisation of adherence towards lipid-lowering therapy are as stated in Table 2.
Conclusion
Patients who have undergone PCI are at a very high risk of developing further CV-related events if their lipid control is suboptimal due to the risk continuum of atherosclerosis. Treatment algorithms pertaining to the usage of lipid-lowering therapy for the purpose of secondary prevention have been well documented in contemporary guidelines. It is imperative that these preventive measures are always adhered to for the overall improvement of patient care and outcomes.
Conflict of interest statement
Z-V Lee has received honoraria from Abbott Vascular, Aspen, AstraZeneca, Bayer, BIOTRONIK, Boehringer Ingelheim, Medtronic, Merck, Merck Sharp & Dohme, Novartis, Pfizer, and Servier. The other author has no conflicts of interest to declare.
