Introduction
Following coronary stent implantation, dual antiplatelet therapy (DAPT) is prescribed to prevent acute and late stent thrombosis and to minimise repeat ischaemic events due to progressive atherosclerosis; however, its use increases the risk of bleeding complications12. In patients not at high bleeding risk, the current European and US guidelines recommend DAPT after percutaneous coronary intervention (PCI) with drug-eluting stents (DES) for at least 6 months in chronic coronary syndromes (CCS) and at least 12 months in acute coronary syndromes (ACS)34, whereas in patients at high bleeding risk, an abbreviated regimen of 1-3 months is suggested5. However, there are currently no recommendations regarding aspirin-free strategies. The history and recent trials of antiplatelet therapy are summarised in Supplementary Appendix 1, Supplementary Table 1 and Supplementary Figure 1–Supplementary Figure 3.
The East Asian paradox and optimal dose of antiplatelet therapy
East Asian patients have a higher prevalence of high on-treatment platelet reactivity compared with Caucasian patients, whilst their thrombotic event rate after PCI is similar, or even lower6. Conversely, the risk of bleeding events in East Asian patients appears to be greater than in Western patients7. This phenomenon, referred to as the “East Asian paradox”, suggests that the optimal therapeutic dose of antiplatelets may be different between East Asian and Western patients (Figure 1).
Based on this hypothesis, a recent clinical trial has shown the efficacy of adjusted-dose prasugrel in Japanese patients8. The multicentre PRASFIT-ACS study in ACS patients showed no significant difference in ischaemic and bleeding events between patients randomised to DAPT followed by low-dose prasugrel (3.75 mg daily) and aspirin (81-330 mg daily) compared to DAPT followed by standard-dose clopidogrel (75 mg daily) and aspirin (81-330 mg daily)8.
Consequently, current guidelines in Japan and Taiwan recommend a reduction in the maintenance dose of prasugrel to 3.75 mg daily after coronary stent deployment910. The current recommended doses of oral antiplatelet therapy after PCI with stent implantation in patients with CCS and non-ST-elevation ACS (NSTE-ACS) are summarised in Table 1. The PENDULUM mono study was a multicentre, non-interventional, prospective registry that assessed the frequency of bleeding complications and cardiovascular events associated with adjusted-dose prasugrel monotherapy (3.75 mg daily) after PCI in high bleeding risk Japanese patients11. In this trial, 65.7% of patients switched to low-dose prasugrel monotherapy at 3 months and up to 83.5% switched at 12 months following their PCI. At 12 months, the cumulative incidence of bleeding complications, defined as Bleeding Academic Research Consortium (BARC) type 2, 3 or 5 after the periprocedural period (from 1 to 12 months after PCI), was 3.2%, and the rate of major adverse cardiac and cerebrovascular events (MACCE), defined as the composite of all-cause death, myocardial infarction (MI), stroke, cerebral infarction, and stent thrombosis, was 3.8%. Furthermore, a prespecified analysis in the PENDULUM mono and the PENDULUM registry comparing single antiplatelet therapy (SAPT) with prasugrel versus DAPT showed that SAPT significantly reduced BARC 2, 3, and 5 bleeding between 0 and 12 months after the index PCI without increasing ischaemic events1112. These observational studies suggest that adjusted-dose prasugrel monotherapy may be safely used in Japanese patients, but this needs to be further demonstrated in prospective studies. In addition, if low-dose prasugrel monotherapy is appropriate for CCS patients, further prospective studies will be needed to explore its feasibility and safety in high-risk patients.
The objective of the present trial is to demonstrate the efficacy and safety of low-dose (3.75 mg daily) prasugrel monotherapy following PCI in Japanese patients presenting with CCS or NSTE-ACS.
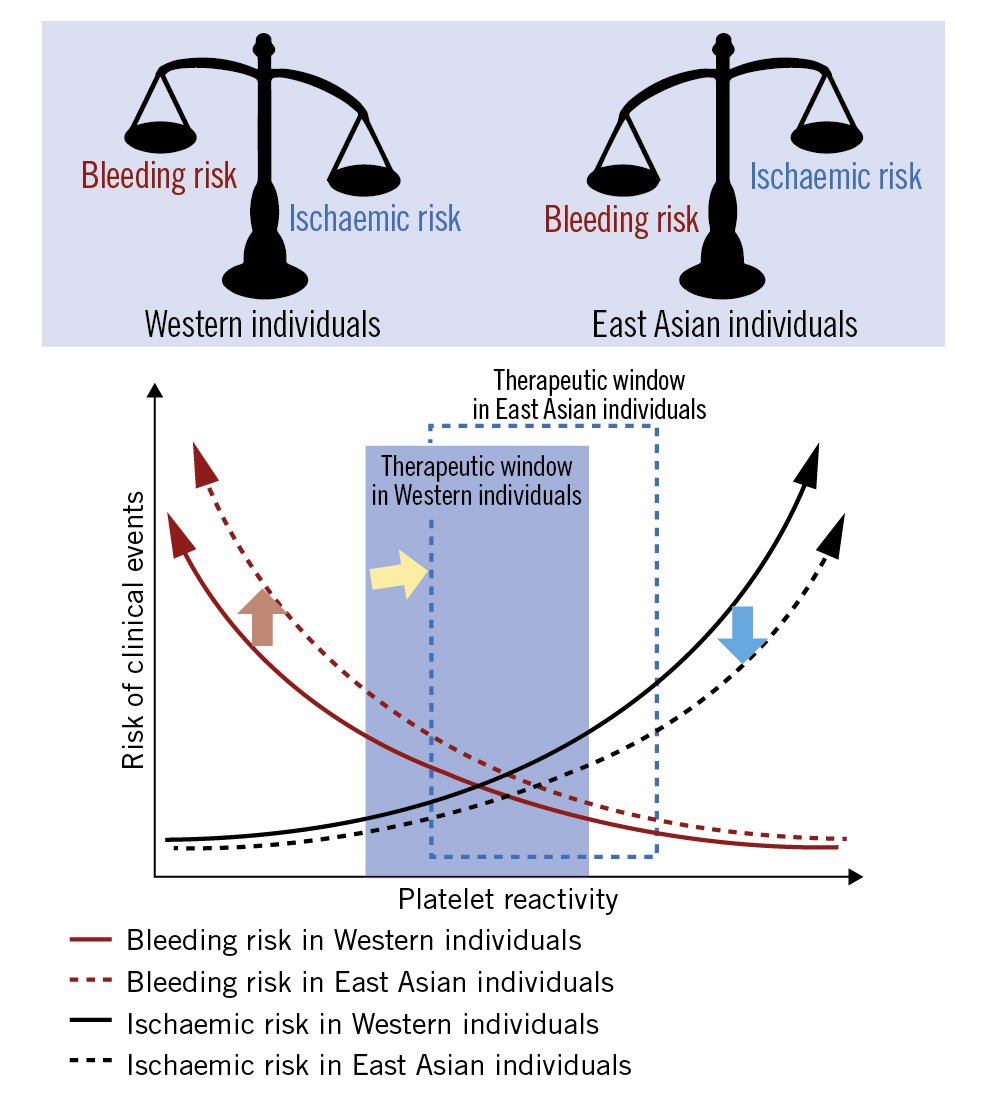
Figure 1. The East Asian paradox. Postulated differences in the optimal “therapeutic window” of platelet reactivity between Western (blue square) and East Asian populations (blue dotted square). Compared to Western individuals, East Asian individuals have lower ischaemic risk at a same level of platelet reactivity (blue arrow); however, bleeding risk in East Asian individuals is higher than Western individuals (red arrow). The optimal “therapeutic window” of platelet reactivity might differ between Western and East Asian patients (yellow arrow).
Table 1. Current guidelines for the dosing of oral antiplatelet therapy after percutaneous coronary intervention with stent implantation due to chronic coronary syndrome and non-ST-elevation acute coronary syndrome.
| Loading dose | Maintenance dose | |||||
|---|---|---|---|---|---|---|
| JCS | ESC | ACC/AHA | JCS | ESC | ACC/AHA | |
| Aspirin | 162-324 mg | 150–300 mg | 162–325 mg | 81–162 mg, once daily | 75–100 mg, once daily | 81 mg, once daily |
| Clopidogrel | 300 mg | 600 mg | 600 mg | 75 mg, once daily | 75 mg, once daily | 75 mg, once daily |
| Prasugrel | 20 mg | **60 mg | †60 mg | 3.75 mg, once daily | ‡10 mg, once daily | ¶10 mg, once daily |
| Ticagrelor | *180 mg | **180 mg | †180 mg | ††90 mg, twice daily | §90 (60) mg, twice daily | ||90 mg, twice daily |
| Reduced doses of P2Y12 inhibitor are highlighted in bold. *Ticagrelor is not available for CCS patients. In patients with NSTE-ACS, a ticagrelor 180 mg loading dose may be considered in P2Y12 receptor inhibitor-naive patients before PCI, when prasugrel or clopidogrel are not available or are contraindicated. **Prasugrel or ticagrelor on top of aspirin may be considered instead of clopidogrel for CCS patients undergoing PCI, considering the ischaemic (e.g., high SYNTAX score, prior stent thrombosis, location and number of implanted stents) and bleeding risks. †Prasugrel and ticagrelor are not available for CCS patients. ††Ticagrelor is considered when prasugrel is not available or contraindicated. ‡5 mg/day for patients aged ≥75 years or with a body weight <60 kg. §Ticagrelor 60 mg is the treatment option for DAPT in combination with aspirin 75–100 mg daily in post-myocardial infarction patients who have tolerated DAPT for 1 year with a high or moderate risk of an ischaemic event, and without a high bleeding risk. ¶In patients with ACS (NSTE-ACS or STEMI) treated with DAPT after coronary stent implantation who are not at high risk for bleeding complications and who do not have a history of stroke or TIA, it is reasonable to choose prasugrel over clopidogrel for maintenance P2Y12 inhibitor therapy. ||In patients with ACS (NSTE-ACS or STEMI) treated with DAPT after coronary stent implantation, it is reasonable to use ticagrelor in preference to clopidogrel for maintenance P2Y12 inhibitor therapy. ACC/AHA: American College of Cardiology/American Heart Association; CCS: chronic coronary syndrome; DAPT: dual antiplatelet therapy; ESC: European Society of Cardiology; JCS: Japanese Circulation Society; NSTE-ACS: non-ST-elevation acute coronary syndrome; PCI: percutaneous coronary intervention; STEMI: ST-elevation myocardial infarction; TIA: transient ischaemic attack | ||||||
Methods
Study design
The Acetyl Salicylic Elimination Trial (ASET) Japan Pilot Study (ClinicalTrials.gov: NCT05117866) is designed as a multicentre, single-arm, open-label, proof-of-concept trial with a stopping rule based on the occurrence of definite stent thrombosis13. The study flowchart is shown in Figure 2. The enrolment of patients across 12 sites in Japan is planned in two phases: 1) 200 patients presenting with CCS, followed by 2) 200 patients presenting with NSTE-ACS. The recruitment will start with patients presenting with CCS, and the Data Safety and Monitoring Board (DSMB) will closely monitor safety events and will meet when recruitment in the CCS cohort reaches 100. The DSMB will then advise the steering committee whether enrolment of the NSTE-ACS population should be started based on clinical events reported at that time. During Phase 1, patient recruitment will be stopped if three or more cases of definite stent thrombosis occur within three months of the index PCI. In Phase 2, enrolment will be stopped if three or more patients experience definite stent thrombosis within 12 months of the index PCI, or if there are two or more sudden deaths in the first 30 days. The cut-off rate for definite stent thrombosis was determined based on the previously reported incidence of stent thrombosis14. In the EVOLVE II Trial, the incidence of stent thrombosis was 0.4% at 30 days and 0.4% at 1 year15, whilst it was 1.2% at 30 days and 1.5% at 1 year in the CARDIOBASE Bern PCI Registry16.
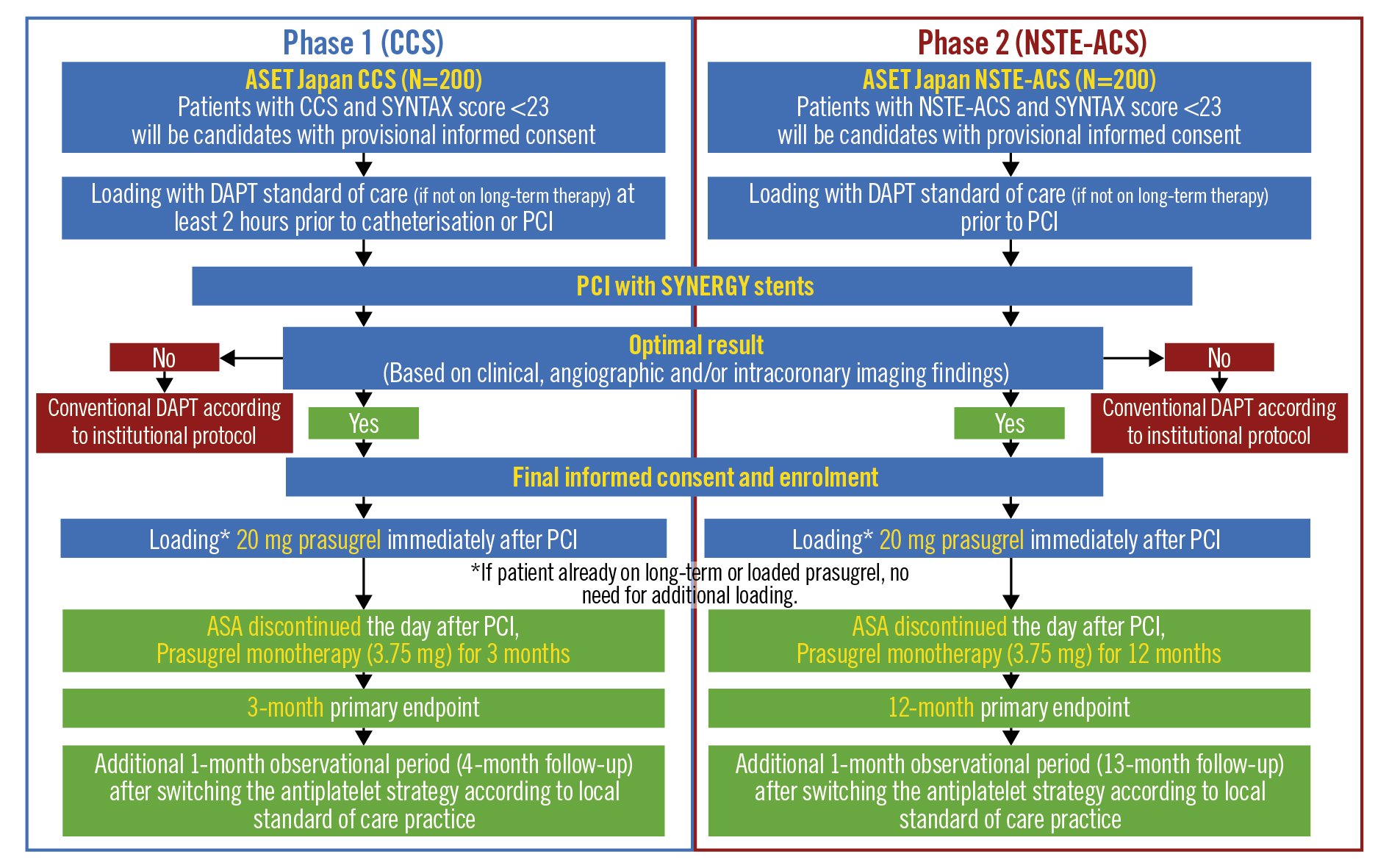
Figure 2. Flowchart of the ASET Japan trial. ASA: acetylsalicylic acid; CCS: chronic coronary syndromes; DAPT: dual antiplatelet therapy; NSTE-ACS: non-ST-elevation acute coronary syndromes; PCI: percutaneous coronary intervention
Sample size considerations
Due to the exploratory nature of this study, no formal sample size calculations were performed. Based on a previous pilot study17, we decided to enrol 200 consecutive patients in each phase (for a of total 400 patients). The Benestent-II Pilot Study, which attempted to eliminate the convention of anti-vitamin K anticoagulation post-stenting that was in use at the time, was the first-in-human study to apply a stopping rule involving a fixed rate of stent thrombosis beyond which the study would have to be terminated. This first-in-human study was successfully completed without any stent thrombosis in 200 patients and was followed by the large randomised Benestent-II Pilot Study, with 827 patients treated without anticoagulation18.
Informed consent
All potential patients provided written informed consent prior to undergoing any study-specific procedures, including screening and diagnostic angiography potentially leading to “ad hoc” PCI. The certified review board (CRB), central ethics committee and local ethics committee at each participating centre approved the study protocol (CR20-023 and CR21-017 at the Fujita Health University). The study protocol has been registered at the Japanese Registry of Clinical Trials (identifier: jRCTs042200053).
Patient screening
Patients requiring PCI for CCS or NSTE-ACS with an anatomical SYNTAX score <23 prior to revascularisation will be screened for enrolment in this trial. The anatomical SYNTAX score will be assessed onsite by trained investigators, and the results will be confirmed by an independent core lab. The inclusion and exclusion criteria are listed in Table 2. Cardiac enzymes and biomarkers must be sampled to detect acute MI prior to the index PCI. The criteria for cardiac enzymes and biomarkers are shown in Supplementary Appendix 2.
Table 2. Inclusion and exclusion criteria of the ASET Japan trial.
| For CCS patients (Phase 1) |
|---|
| 1. Inclusion criteria for CCS patients |
| All candidates must meet the following inclusion criteria: |
| 1. Successful PCI with optimal acute stent implantation of one or more SYNERGY stent(s). |
| 2. SYNERGY stent implantation was performed to treat: |
| a) at least one de novo lesion with ≥50% diameter stenosis determined by visual assessment in at least one native coronary artery with a vessel size between 2.25 mm and 5.0 mm in diameter. |
| b) non-acute coronary disease, with normal cardiac biomarker values prior to the PCI procedure, and evidence of myocardial ischaemia by symptoms or non-invasive/invasive testing. |
| c) patients with anatomical SYNTAX score <23 prior to PCI |
| 3. Patient has provided written informed consent as approved by the ethics committee of the respective clinical site. |
| 2. Exclusion criteria for CCS patients |
| Candidates will be ineligible for enrolment if any of the following conditions apply: |
| 1. ≤20 years of age. |
| 2. Unable to give informed consent. |
| 3. Females of child-bearing potential unless negative pregnancy test at screening and willing to use effective contraception for the duration of treatment with study medication. |
| 4. Females who are breastfeeding at time of enrolment. |
| 5. Patients concomitantly receiving any other non-study stent for the same procedure. |
| 6. Patients with planned PCI or surgical intervention to treat any cardiac or non-cardiac condition. |
| 7. Previous PCI with any non-SYNERGY stents in the last 6 months. |
| 8. Current (same hospitalisation) or previous (within 12 months) acute coronary syndrome. |
| 9. Patients with the following lesion characteristics prior to PCI: saphenous or arterial graft, in-stent (re)stenosis. |
| 10. History of definite stent thrombosis. |
| 11. Concomitant cardiac valve disease requiring invasive therapy. |
| 12. Atrial fibrillation or other indication for oral anticoagulant therapy. |
| 13. Known allergy to aspirin or prasugrel or diagnosed lactose intolerance. |
| 14. Acute heart failure. |
| 15. Active myocarditis. |
| 16. Cardiomyopathy. |
| 17. Patient in haemodialysis. |
| 18. Treatment in the last 10 days or requirement for ongoing treatment with a strong CYP3A4 inhibitor or inducer. |
| 19. History of stroke or transient ischaemic cerebrovascular accident. |
| 20. History of intracranial haemorrhage or other intracranial pathology associated with increased bleeding risk. |
| 21. Haemoglobin <10 g/dL or other evidence of active bleeding. |
| 22. Peptic ulceration documented by endoscopy within the last 3 months unless healing proven by repeat endoscopy. |
| 23. Any other condition deemed by the investigator to place the patient at excessive risk of bleeding with prasugrel. |
| 24. Participation in another trial with an investigational drug or device. |
| 25. Comorbidity associated with life expectancy <1 year. |
| 26. Assessment that the subject is not likely to comply with the study procedures or have complete follow-up. |
| 27. Known drug or alcohol dependence within the past 12 months as judged by the investigator. |
| CCS: chronic coronary syndrome; PCI: percutaneous coronary intervention |
| For NSTE-ACS patients (Phase 2) |
| 1. Inclusion criteria |
| For NSTE-ACS patients |
| 1. Patients with diagnosed non-ST-elevation acute coronary syndrome. |
| 2. Patients with anatomical SYNTAX score <23 prior to PCI. |
| 3. Patient provided written informed consent as approved by the ethics committee of the respective clinical site. |
| Post-PCI for NSTE-ACS patients |
| 1. Patient is free of angina symptoms at the end of PCI procedure. |
| 2. Successful PCI with optimal acute stent implantation of one or more SYNERGY stent(s). |
| 3. SYNERGY stent implantation was performed to treat at least one de novo lesion with ≥50% diameter stenosis determined by visual assessment in at least one native coronary artery with a vessel size between 2.25 mm and 5.0 mm in diameter. |
| 2. Exclusion criteria |
| Candidates will be ineligible for enrolment if any of the following conditions apply: |
| 1. ≤20 years of age. |
| 2. Unable to give informed consent. |
| 3. Females of child-bearing potential unless negative pregnancy test at screening and willing to use effective contraception for the duration of treatment with study medication. |
| 4. Females who are breastfeeding at time of enrolment. |
| 5. Patients concomitantly received any other non-study stent at the same procedure. |
| 6. Patients with planned PCI or surgical intervention to treat any cardiac or non-cardiac condition. |
| 7. Previous PCI with any non-SYNERGY stents in the last 6 months. |
| 8. Patient with following lesion characteristics prior to PCI. |
| 9. History of definite stent thrombosis. |
| 10. Concomitant cardiac valve disease requiring invasive therapy. |
| 11. Known allergy to aspirin, prasugrel or diagnosed lactose intolerance. |
| 12. Atrial fibrillation or other indication for oral anticoagulant therapy. |
| 13. History of stroke or transient ischaemic cerebrovascular accident. |
| 14. History of intracranial haemorrhage or other intracranial pathology associated with increased bleeding risk. |
| 15. Acute heart failure. |
| 16. Active myocarditis. |
| 17. Cardiomyopathy. |
| 18. Patient in haemodialysis. |
| 19. Haemoglobin <10 g/dL or other evidence of active bleeding. |
| 20. Haemodynamic instability or cardiogenic shock. |
| 21. Recurrent or ongoing chest pain refractory to medical treatment. |
| 22. Life-threatening arrhythmias or cardiac arrest. |
| 23. Mechanical complications of myocardial infarction. |
| 24. Recurrent dynamic ST-T wave changes, particularly with intermittent ST-elevation. |
| 25. Peptic ulceration documented by endoscopy within the last 3 months unless healing proven by repeat endoscopy. |
| 26. Any other condition deemed by the investigator to place the patient at excessive risk of bleeding with prasugrel. |
| 27. Participation in another trial with an investigational drug or device. |
| 28. Comorbidity associated with life expectancy <1 year. |
| 29. Assessment that the subject is not likely to comply with the study procedures or have complete follow-up. |
| 30. Known drug or alcohol dependence within the past 12 months as judged by the investigator. |
| ACS: acute coronary syndrome; NSTE-ACS: non-ST-elevation acute coronary syndrome; PCI: percutaneous coronary intervention |
PCI procedure
All patients will be loaded with standard DAPT (aspirin 81 to 330 mg and clopidogrel 300 mg, prasugrel 20 mg, or ticagrelor 180 mg, unless patients are on long-term [≥5 days prior to the index PCI] therapy with prasugrel 3.75 mg as a Japanese standard maintenance dose), which according to local practice is at least two hours prior to their index PCI for CCS patients (Phase 1), and prior to their PCI procedure for NSTE-ACS patients (Phase 2). The use of glycoprotein IIb/IIIa inhibitors is regulated in Japan by the Pharmaceuticals and Medical Devices Agency (PMDA), and previous trials have demonstrated that abciximab does not reduce major coronary events, whilst significantly increasing bleeding and thrombocytopaenia19.
The index PCI will be performed using a radial, brachial or femoral approach with the intention to achieve complete revascularisation in at least one stenosis with an angiographic diameter stenosis ≥50%, as identified by the local interventional cardiologist. Although not mandated, the radial approach will be strongly recommended. Periprocedural anticoagulation will be used at the operator’s discretion according to local or international guidelines20. All target lesions must be exclusively treated with the SYNERGY stent (Boston Scientific), which elutes everolimus within three months from a 4 μm biodegradable poly(lactic-co-glycolic acid) (PLGA) coating that is located only on the abluminal side of 74 μm/79 μm/81 μm platinum-chromium struts (for the stent sizes ≤2.5 mm/3.0-3.5 mm/4.0 mm, respectively) and is resorbed within four months. These features have been introduced to improve early endothelialisation of stent struts, accelerate vessel healing, and reduce thrombotic events. The SYNERGY stent has been shown to have clinical safety and efficacy in a previous randomised trial15. The feasibility and safety of the SYNERGY stent with prasugrel monotherapy without aspirin were shown in the ASET study17. The potential advantage of uniform use of the SYNERGY stent is to facilitate the pooling of data with that of the ASET study in which the SYNERGY stent was also uniformly used. However, the potential disadvantage of this is that evidence stemming from this study will not be generalisable to other technologies.
The investigator should perform the procedure to achieve optimal stent implantation according to local standards of care by angiography including quantitative coronary angiography (QCA) and/or findings from intracoronary imaging (intravascular ultrasound [IVUS], optical coherence tomography [OCT], or optical frequency domain imaging [OFDI]). An optimal immediate coronary stent result is a combination of successful stent implantation with no significant residual diameter stenosis (<20%), no major edge dissection, no angiographic finding suggestive of thrombus, no major side branch occlusion, no post-procedure delay in contrast filling, and no major stent incomplete apposition21. Use of intracoronary imaging pre- and/or post-stent implantation for optimisation is left to the operator’s discretion. After the index PCI, only if the angiographic and/or intravascular results are considered satisfactory in the operator’s clinical judgement will the patient be enrolled in the study.
In cases of screening failure, the reasons why optimal stent implantation was not achieved need to be recorded.
Enrolment and follow-up
After achievement of optimal SYNERGY stent implantation, patients will be enrolled in the study and loaded with prasugrel 20 mg immediately in the cath lab to avoid further delays in loading and maintained on prasugrel 3.75 mg monotherapy once daily for 3 months in Phase 1, and for 12 months in Phase 2. This strategy of switching P2Y12 inhibitors is in line with the international consensus on switching therapies and is supported by pharmacodynamic studies2223. Patients who were loaded with prasugrel preprocedure or have been on long-term prasugrel do not need additional loading. Aspirin will be discontinued the day after the index PCI.
In Phase 1, clinical follow-up with outpatient visits will be performed at 3 months and with telephone contacts at 1 and 4 months. In Phase 2, clinical follow-up will be performed with outpatient visits at 1 and 12 months and telephone contact at 3, 6, and 13 months. The 4- and 13-month telephone follow-ups are not related to the scientific purpose of this pilot study; this period is rather for observational assessment after switching the standard of care (aspirin alone, P2Y12 inhibitor monotherapy, or DAPT). An assessment of angina status, cardiovascular drug use, and any serious adverse events will be recorded during clinical follow-up visits.
Optimal medical therapy with strict control of low-density lipoprotein cholesterol is strongly recommended along with optimisation of all medication therapy according to current guidelines20. The use of other medications (e.g., beta blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers) will also be recommended.
Study endpoints
The study endpoints are described in Table 3. The primary endpoint is a composite of cardiac death, target vessel MI (>48 hours post-PCI), defined as an MI case with evidence of myocardial necrosis in the vascular territory of a previously treated target vessel, or definite stent thrombosis occurring within 3 months of the index PCI in CCS patients or 12 months for NSTE-ACS patients. Inclusion in this trial occurs after the index procedure; hence, only MI occurring >48 hours after the index PCI will be taken into consideration as a primary endpoint. The primary bleeding endpoint is any BARC type 3 or 5 bleeding occurring within 3 months of the index PCI in CCS patients or 12 months for NSTE-ACS patients.
Secondary endpoints will include all-cause death, stroke (subclassified as ischaemic, haemorrhagic, or unknown), all MI, repeat revascularisation, definite/probable/possible stent thrombosis, BARC type 1-5 bleeding, and each individual component of the primary endpoint. The patient-oriented composite endpoint (PoCE) is defined as a composite of all-cause death, any stroke, any MI, or revascularisation. The device-oriented composite endpoint (DoCE) is defined as a composite of cardiovascular death, target vessel MI, or clinically driven target lesion revascularisation. Net clinical adverse events are defined as a composite of PoCE and BARC type 3 or 5 bleeding.
All deaths will be considered cardiac unless an undisputed non-cardiac cause can be established. Spontaneous MI will be defined according to the Fourth Universal definitions24. Periprocedural MI (<48 hours post-PCI) will be defined according to the Society for Cardiovascular Angiography and Interventions (SCAI) 2013 definition25. Stent thrombosis will be defined according to the Academic Research Consortium (ARC)-2 definition13. BARC bleeding will be defined as previously reported26. Safety measures will assess any drug discontinuation rate and date.
Table 3. Endpoints of the ASET Japan trial.
| Study endpoints. | |
|---|---|
| Primary ischaemic endpoints | |
| A composite of cardiac death, target vessel myocardial infarction (>48 hours post-PCI), and definite stent thrombosis up to 3 months for CCS patients (Phase 1) and up to 12 months for NSTE-ACS patients (Phase 2) after the index procedure. | |
| Primary bleeding endpoints | |
| The primary bleeding endpoint is any BARC type 3 or 5 bleeding up to 3 months for CCS patients, and 12 months for NSTE-ACS patients after the index PCI. | |
| Secondary endpoints | |
| 1. All-cause death. | |
| 2. Stroke | Ischaemic |
| Haemorrhagic | |
| Unknown | |
| 3. All myocardial infarctions. | |
| 4. Repeat revascularisation. | |
| 5. Definite/probable/possible stent thrombosis. | |
| 6. BARC type 1–5 bleedings and each individual component of the primary endpoint. | |
| 7. Patient-oriented composite endpoints (PoCE). | |
| 8. Device-oriented composite endpoints (DoCE). | |
| 9. Net adverse clinical event (NACE). | |
| ACS: acute coronary syndrome; BARC: Bleeding Academic Research Consortium; CCS: chronic coronary syndrome; DoCE: device-oriented composite endpoint; NACE: net adverse clinical event; NSTE ACS: non-ST-elevation acute coronary syndrome; PCI: percutaneous coronary intervention; PoCE: patient-oriented composite events | |
Clinical events adjudication and data safety monitoring
All events will be adjudicated by an independent clinical events committee. An independent DSMB will monitor the safety and efficacy of all the patients during enrolment and 3-month follow-up in CCS patients, and during enrolment and 12-month follow-up in NSTE-ACS patients, including the stopping rule based on the occurrence of definite stent thrombosis.
Discussion
Several randomised controlled trials have demonstrated the efficacy of potent P2Y12 receptor inhibitor monotherapy following 1- or 3-month DAPT in CCS and ACS patients treated by PCI27282930. The characteristics of contemporary clinical trials of ticagrelor or clopidogrel monotherapy after PCI are summarised in Supplementary Table 1. The first-in-human ASET study was conducted in Brazil as a multicentre, single-arm, open-label trial and demonstrated the feasibility and safety of aspirin-free prasugrel monotherapy following PCI using the SYNERGY stent in low-risk patients with CCS or stabilised ACS17. Among the 201 enrolled patients, no stent thrombosis occurred with a high adherence to prasugrel (98.3%) during the 4-month follow-up period. The primary bleeding endpoint, defined as BARC type 3 or 5 bleeding, occurred in one patient (0.5%). Currently at least 3 clinical trials, STOPDAPT-3, NEOMINDSET, and OPTICA, are exploring novel strategies of monotherapy using P2Y12 receptor inhibitors (Supplementary Table 2).
Limitations
With regard to limitations of the present study, firstly, the findings need to be considered as hypothesis-generating due to the single-arm design with a small sample size without formal sample size calculation. Secondly, only patients with optimal angiographic and/or intravascular results will be enrolled in the study. This is a potential issue in terms of generalising the ASET Japan results into practice. Finally, due to the protocol of the trial, some of the participants will be required to have a loading dose of P2Y12 inhibitor twice. Although this strategy has been accepted in line with expert consensus, it potentially could affect the results.
Conclusions
With this “no DAPT” study, the investigators expect to demonstrate the feasibility and safety of prasugrel monotherapy just after stent deployment in selected Japanese patients with CCS and NSTE-ACS. Further randomised controlled trials are needed to evaluate the aspirin-free strategy compared with traditional DAPT following PCI.
Funding
The ASET Japan is an investigator initiated study, and Meditrix Corporation is the sponsor. This trial is supported by grants from Boston Scientific Japan. The authors declare this funding source had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Conflict of interest statement
S. Masuda reports a grant from Terumo outside the submitted work. T. Muramatsu has received honoraria/speaker fees from Boston Scientific Japan and Daiichi Sankyo. K. Kozuma received honoraria for lectures and is a member of the advisory boards of Daiichi Sankyo and Boston Scientific. K. Tanabe received honoraria from Boston Scientific and Daiichi Sankyo. M. Nakamura reports grants from Daiichi Sankyo during the conduct of the study; and honoraria from Bayer KK, Daiichi Sankyo KK, and Japan Lifeline. Y. Morino received a scientific grant and lecture fee from Boston Scientific. P.W. Serruys reports institutional grants from Sino Medical Sciences Technology, SMT, Philips Volcano, Xeltis, and HeartFlow, outside the submitted work. The other authors have no conflicts of interest to declare.
